Definición
El síndrome clásico
de Guillain-Barré incluye todas aquellas entidades clínicas caracterizadas por
la afectación aguda o subaguda de las raíces y nervios periféricos debidas a un
mecanismo inmunomediado (poliradiculoneuropatía aguda de origen autoinmune).
Estas entidades, sintetizadas bajo un epónimo común, se presentan con diferentes
manifestaciones clínicas y características electrofisiológicas.
Etiología
El síndrome de
Guillain-Barré es la causa más frecuente de polineuropatía aguda en nuestro
medio y constituye la primera causa de parálisis aguda de origen neuromuscular en
países occidentales. Puede aparecer en todos los grupos de edad, aunque es poco
frecuente en la infancia. Hay antecedentes de infección respiratoria o
intestinal en las dos semanas previas en 2/3 de los pacientes, por lo que se
cree que determinados virus o bacterias juegan un papel predisponente. El
patógeno que se ha relacionado con más frecuencia es el Campylobacter jejuni
(20% de los casos estudiados).
Existen casos
relacionados con vacunaciones contra la gripe estacional y la gripe A.
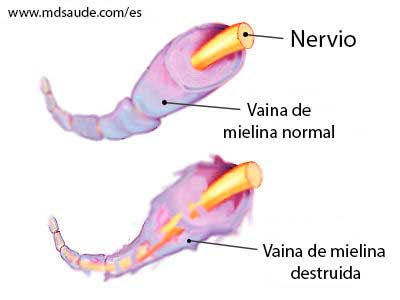
Clasificación
En base a aspectos
clínicos y electrofisiológicos, podemos diferenciar tres subtipos de síndrome
de Guillain-Barré:
1) la
poliradiculopatía desmielinizante inflamatoria aguda (AIDP), que constituye la
forma clínica más frecuente en nuestro medio; y las formas axonales, en las que
se incluyen;
2) la neuropatía
axonal motora aguda (AMAN).
3) la neuropatía
axonal motora y sensitiva aguda (AMSAN). Las formas axonales son mucho más
frecuentes en los países orientales (< 5% en nuestro medio).
Existen variantes topográficas
del síndrome de Guillain-Barré, poco frecuentes: el síndrome de Miller-Fisher
(caracterizado por oftalmoplejía, ataxia y arreflexia), la parálisis
faringocérvico-braquial (marcada debilidad de la musculatura orofaríngea, de
hombros y cuello), la diplejía facial con parestesias distales o la
pandisautonomía pura.
Manifestaciones
clínicas
Cursa clínicamente
con una parálisis muscular simétrica de inicio agudo que se inicia distalmente
y asciende proximalmente durante horas o días hacia regiones no afectadas
inicialmente. La debilidad puede incluir la musculatura facial de forma bilateral
(hasta en el 50% de los pacientes) y, menos frecuentemente, la extraocular.
Junto con la debilidad muscular, aparece hipo o arreflexia (no es excepcional
que los reflejos estén preservados, especialmente en fases iniciales).
Alteraciones sensitivas, en la mayoría de los casos en forma de parestesias y/o
disestesias en las zonas acras.
Sin embargo, la
presencia de hipoestesia debe hacer que nos replanteemos el diagnóstico.
Un 25% de pacientes
presentan dolor punzante en la espalda o una extremidad que, además, puede
preceder al resto del cuadro clínico. Puede existir disfunción autonómica, que
suele ser subclínica, o bien aparecer en forma de taquicardia y alteraciones de
la tensión arterial (hipotensión o hipertensión). La afectación de la función vesical
es muy rara y debe hacernos pensar en otra causa. Los nervios frénicos pueden verse
afectados, llevando a fallo de la musculatura respiratoria.

Diagnóstico
Criterios de
diagnóstico: el diagnóstico es eminentemente clínico, apoyado en el examen del
líquido cefalorraquídeo (LCR) y en el estudio neurofisiológico. Los criterios diagnósticos
de Asbury y Cornblath siguen siendo los más utilizados en la práctica clínica.
Criterios
necesarios para el diagnóstico
1. Debilidad motora
progresiva de más de un miembro
2. Arreflexia o
hiporreflexia marcada (en ocasiones los reflejos pueden estar conservados, especialmente
en las fases iniciales)
Datos que apoyan
el diagnóstico
1. Progresión a lo
largo de días o semanas (el 80% se produce en dos semanas)
2. Relativa
simetría
3. Pérdida leve de
la sensibilidad
4. Comienzo con
dolor o malestar de una extremidad
5. Compromiso de
nervios craneales
6. Comienzo de la
recuperación a las 2-4 semanas tras detenerse la progresión
7. Trastorno
funcional autonómico
8. Ausencia de
fiebre al comienzo de la evolución
9. Aumento en el
nivel de las proteínas del LCR una semana después de la aparición de los
síntomas
10. Hallazgos
electrofisiológicos típicos
Datos que ponen
en duda el diagnóstico
1. Nivel sensitivo
2. Asimetría
marcada y persistente
3. Disfunción
vesical o intestinal persistente
4. Más de 50
células/mm3 en el LCR
Pruebas complementarias:
Análisis de sangre:
por lo general los estudios rutinarios de hemograma, bioquímica y coagulación
son normales excepto en el caso de que el cuadro se haya precedido de una
infección reciente, en este caso habrá alteraciones relacionadas con este proceso.
Análisis de líquido
cefalorraquídeo: hasta el 95% de los pacientes presentan disociación albúmino-citológica
(elevación del número de proteínas sin pleocitosis acompañante) en el LCR, que
aparece a partir del séptimo día de evolución.
Estudios
electrofisiológicos: los estudios de conducciones nerviosas son la prueba de
mayor utilidad. Es típico un enlentecimiento generalizado en las velocidades de
conducción. En los 4-6 primeros días los hallazgos pueden ser inespecíficos o
normales.
Tratamiento
Medidas
generales.
En todos los
pacientes se debe monitorizar la función respiratoria, cardiaca y hemodinámica.
Vigilar los signos y los síntomas que predicen fallo respiratorio: diaforesis,
alteración de la consciencia, uso de los músculos accesorios, hipoxemia o
hipercapnia en la gasometría… El tratamiento debe centrarse, si es necesario, en
el soporte ventilatorio, manejo del dolor (frecuentes cambios de posición y
analgesia), de las alteraciones disautonómicas (útil el propranolol 1
mg/kg/día) y un manejo nutricional específico (la vía enteral es de elección;
la dieta debe ser hiperproteica e hipercalórica debido a su estado
hipercatabólico).
Tratamiento
específico
La inmunoterapia
(inmunoglobulinas intravenosas y plasmaféresis) constituye la piedra angular
del tratamiento específico y debe administrase precozmente. Las
inmunoglobulinas intravenosas se administran en dosis total de 2 g/kg que, por
lo general, se divide en dosis de 400 mg/kg/día durante 5 días (se pueden
administrar dosis adicionales si existe recaída). El volumen total de plasma que
se recambia en la plasmaféresis no está bien establecido, pero suele ser de
entre 200-250ml/Kg durante 7 a 14 días. Si existira una recaída en los días
próximos a la realización de la plasmaféresis se pueden realizar sesiones
adicionales. La combinación de tratamiento alternando plasmaféresis e
inmunoglobulinas intravenosas no ha demostrado ningún beneficio. Tampoco ha
demostrado utilidad el uso aislado o en combinación de corticoides. En las
formas leves, capaces de caminar, no hay consenso acerca de si deben o no ser
tratadas con inmunoterapia.
Destino
del Paciente
El paciente ha de
ser hospitalizado siempre, incluso en los casos con afectación mínima. La gran
mayoría de los pacientes que requieren tratamiento suelen ser tratados en UCI.
OTRAS
POLINEUROPATÍAS AGUDAS
Polineuropatía
alcohólica:
aparece en pacientes con una ingesta de alcohol intensa y mantenida durante un
tiempo prolongado. Presentan polineuropatía sensitivo motora simétrica distal.
El curso es habitualmente crónico, aunque se han descrito episodios de
empeoramiento agudo o subagudo que hay que diferenciar del síndrome de
Guillain-Barré.
Polineuropatía
VIH: Durante la
seroconversión puede aparecer una polineuropatía desmielinizante aguda
indiferenciable del síndrome de Guillain-Barré. El LCR muestra elevación de las
proteínas, pero con mayor celularidad (por encima de 30 células/mm3).
Polineuropatía de
la enfermedad de Lyme:
La enfermedad de Lyme, producida por la infección por Borrelia burgdorferi,
puede presentarse en forma de una meningoradiculitis subaguda, con afectación
frecuente del nervio facial. El LCR muestra pleocitosis mononuclear e
hiperproteinoraquia.
Polineuropatías
tóxicas: La
polineuropatía por arsénico se manifiesta en forma de disestesias dolorosas que
afectan primero a los pies y luego a las manos. Cursa con debilidad muscular de
predomino distal y pérdida de reflejos. A diferencia del síndrome de
Guillain-Barré, el déficit propioceptivo suele ser grave, con ataxia
importante.

No hay comentarios:
Publicar un comentario